Dans son travail, le chercheur ou la chercheuse en biologie peut être confronté à des spécimens biologiques, qu’il s’agisse d’individus entiers ou d’échantillons (os, fèces…), pour lesquels il ou elle n’est pas capable de proposer une détermination. Cela peut venir de connaissances incomplètes en taxonomie, de difficultés particulières propres à l’espèce (dimorphisme sexuel, espèces sœurs…), ou du fait qu’il s’agisse d’un fragment limité de l’individu. Dans ces cas de figure, les codes-barres ADN peuvent être utilisés pour proposer une identification. Cette technique repose sur l’idée que, en séquençant un fragment d’ADN du spécimen inconnu et en le comparant à une base de données de référence, il est possible de l’identifier. Des millions de codes-barres sont disponibles, représentant plusieurs centaines de milliers d’espèces. Au-delà des nombreuses applications pratiques, cette méthode a entraîné un renouveau de la taxonomie, la science qui décrit la biodiversité. Cette dernière reste encore largement inconnue et menacée d’extinction.
En 2003, le Canadien Paul Hebert propose une nouvelle approche d’identification des spécimens reposant sur l’utilisation de codes-barres ADN (DNA barcodes) 12. Cette méthode, appelée DNA barcoding, va rapidement connaître un succès retentissant. À partir d’un fragment d’ADN, il serait possible d’identifier n’importe quel organisme vivant, c’est-à-dire de l’associer à un nom d’espèce. Cette méthode permettrait de s’affranchir des problèmes liés au manque de taxonomistes, capables d’identifier des spécimens sur la base de leurs caractères morphologiques, ou encore à la variabilité morphologique, parfois difficilement interprétable au sein d’une espèce.
Le séquençage de l’ADN est possible depuis la fin des années 70, grâce à la technique dite de Sanger. Cette technique valut le prix Nobel de chimie en 1980 à son inventeur, le Britannique Frederick Sanger. Malgré son rendement faible, cette méthode reste largement utilisée de nos jours, même si depuis d’autres ont vu le jour, comme les méthodes de séquençage de nouvelle génération (next-generation sequencing, NGS) dans les années 2000 et les technologies dites de troisième génération (TGS, third generation sequencing) ou de lectures longues (long reads) dans les années 2010. Ces dernières permettent de séquencer une quantité d’ADN (mesurée en nombre de paires de bases) bien plus importante, pour un coût/base bien plus faible, mais restent plus difficiles à mettre en œuvre.

Dès les années 80, les scientifiques ont bien évidemment utilisé l’ADN, et les séquences qui commençaient à être disponibles, pour répondre à des questions d’évolution, et notamment de taxonomie. Cette science a pour objet de délimiter et décrire les taxons, ainsi que de fournir les outils pour les reconnaître 3. D’ailleurs, en 1993, Arnot et collaborateurs avaient déjà proposé l’idée d’utiliser des codes-barres ADN 4, sans pour autant connaître le succès qu’aura l’idée de Paul Hebert dix ans plus tard. Outre une stratégie de communication plus efficace, Hebert, en bon visionnaire, a directement pensé à associer au principe du DNA barcoding les notions de changement d’échelle et d’universalité.
1. Un changement d’échelle : appliquer le DNA barcoding à l’ensemble du vivant
Plutôt que de se limiter à l’étude d’un taxon en particulier, ce que ses prédécesseurs avaient fait jusqu’à présent, Hebert a proposé dans ses deux articles fondateurs ( 1 et 2) d’appliquer le principe du DNA barcoding à l’ensemble des êtres vivants. Autrement dit, il propose la construction d’une base de données de références, qui contiendrait des codes-barres ADN pour l’ensemble des espèces vivantes sur Terre. Une fois cette base de données complétée, il suffirait donc, pour identifier un être vivant, de prélever son ADN, de séquencer le locus servant à son identification (voir partie suivante), de le comparer à la base de données, d’y trouver la séquence la plus ressemblante, et d’en déduire à quelle espèce appartient l’organisme échantillonné.
Depuis 2003, de nombreux projets, d’abord locaux, puis rapidement internationaux, ont vu le jour pour compléter des bases de données de référence, avec par exemple le consortium iBOL, et les projets Barcode 500 K et Bioscan. Début 2025, la page d’accueil du site web BOLD (Barcode Of Life Datasystem, Figure 1), qui héberge la base de données de référence, annonce un total de plus de 20 millions de codes-barres disponibles, représentant plus d’un million d’espèces, dont un tiers seulement correspond à des espèces décrites, c’est-à-dire ayant fait l’objet d’une publication, avec un nom scientifique et une description de l’espèce. Le chemin reste encore long pour couvrir l’ensemble de la diversité spécifique (plus de 2 millions d’espèces décrites, pour un total estimé aux alentours de 7 à 10 millions d’espèces 5). Cependant, lentement mais sûrement, la base se complète, grâce aux projets de séquençage de plus en plus nombreux, qu’ils concernent des spécimens collectés lors de missions de terrain, comme celles organisées dans le cadre du programme d’expéditions « La Planète Revisitée » du Muséum national d’Histoire naturelle de Paris, ou qu’ils valorisent les spécimens déjà présents dans les collections des muséums.
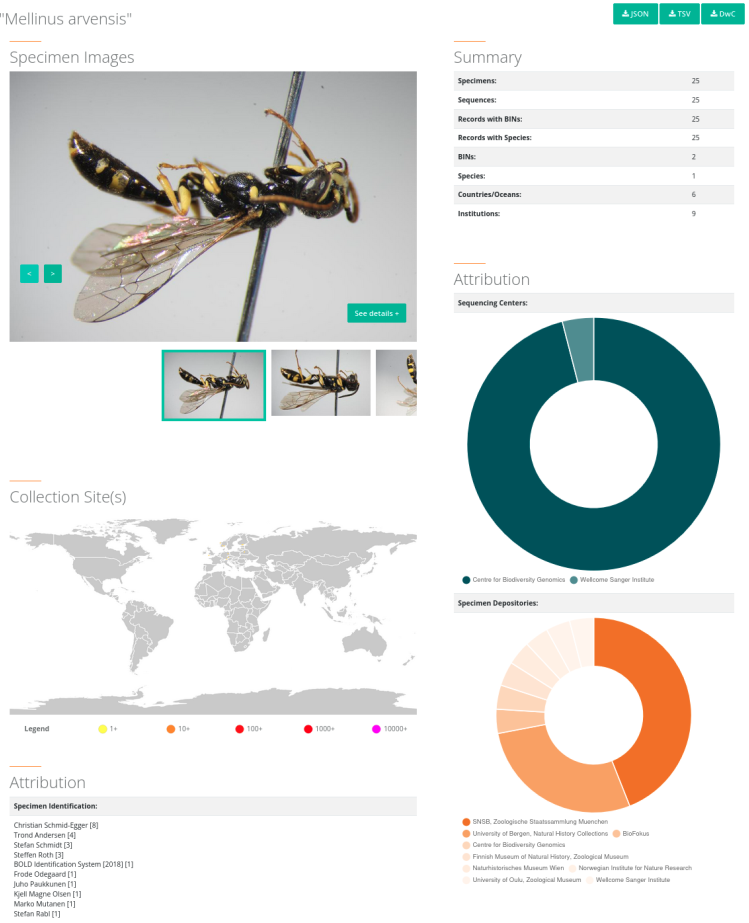
Le site Barcode of Life Data Systems (BOLD) recense les codes-barres associés à différents spécimens de nombreuses espèces. Chaque page consacrée à une espèce (ici Mellinus arvensis) contient notamment des photographies des spécimens, l'origine géographique de ceux-ci, les noms des personnes ou entités ayant déposé les séquences et, bien entendu, des liens d'accès aux codes-barres eux-mêmes.
2. Une universalité relative
Hebert et ses collaborateurs ont proposé d’utiliser comme code-barre universel le gène COI, (ou, selon la nomenclature internationale, cox-1) qui, codant la sous-unité I de la cytochrome oxydase, a l’avantage d’être présent chez tous les Eucaryotes. De plus, il est généralement facile à séquencer, grâce à des amorces universelles servant aussi bien à l’amplification (PCR) qu’au séquençage, et parce que le génome mitochondrial est présent en de nombreux exemplaires dans chaque cellule. Mais surtout, ce fragment d’ADN d’environ 600 paires de bases présente un niveau de variabilité qui en fait un bon code-barres : les différences entre les séquences de ce gène chez différents individus, correspondant aux mutations apparues au cours du temps, sont généralement faibles entre les individus d’une même espèce et plus élevées entre des individus d’espèces différentes. C’est ainsi que cette séquence d’ADN peut être employée, à l’instar des codes-barres utilisés dans le commerce, comme un outil d’identification taxonomique : tous les individus d’une même espèce ont des séquences relativement similaires, ce qui permet l’identification d’un spécimen en comparant sa séquence avec celles des espèces connues.
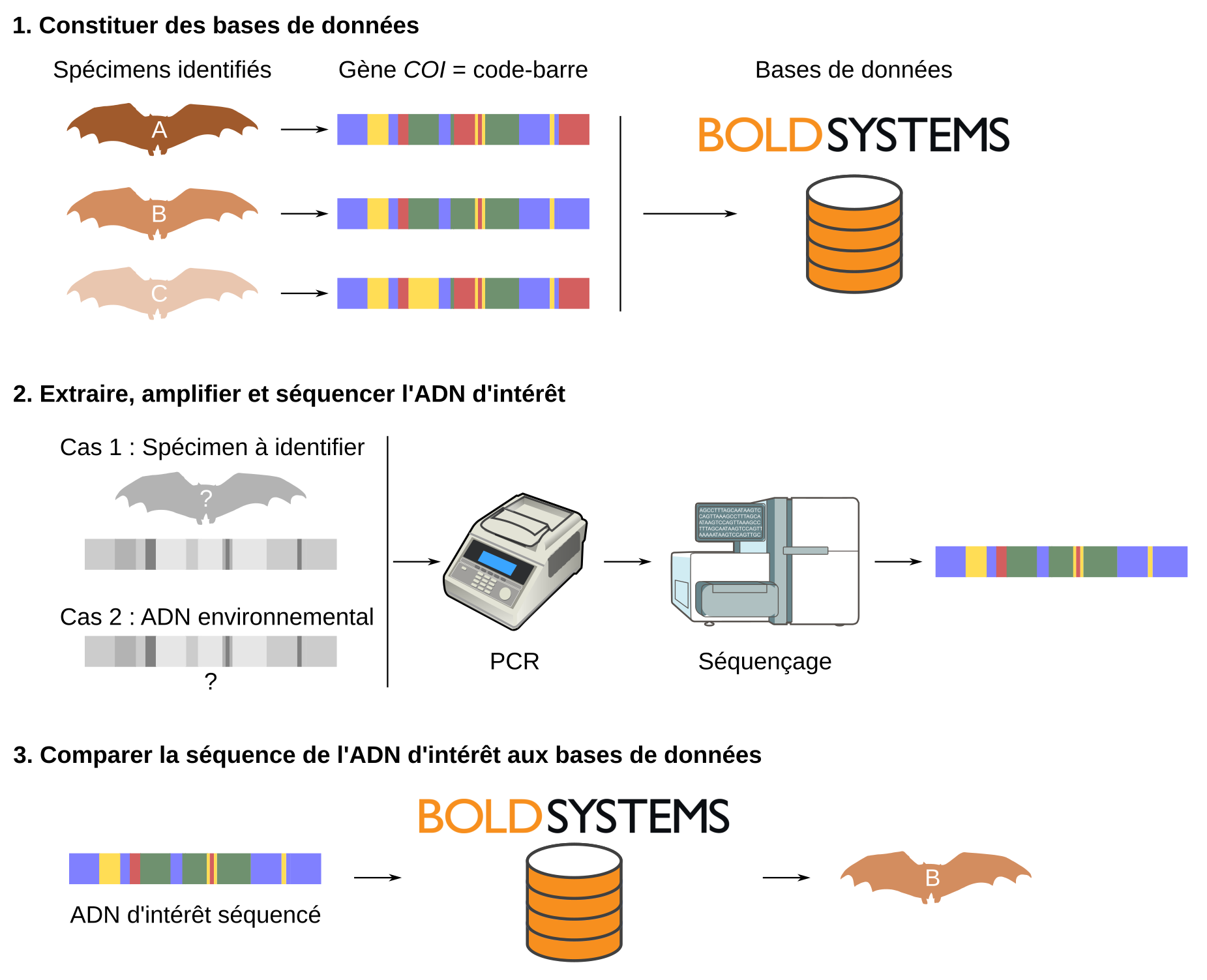
1. Pour pouvoir identifier des spécimens à partir de leur ADN, il est nécessaire de construire au préalable une base de données (comme la Barcode of Life Database, BOLD) dans laquelle des spécimens sont associés à des noms d’espèces et à un code-barre ADN. Chez les animaux, le code-barre utilisé est le gène mitochondrial COI, d’environ 600 paires de bases. En effet, ce gène présente une grande variabilité interspécifique, mais une faible variabilité intraspécifique. D’autres gènes peuvent être utilisés selon les taxons.
2. À l’étape suivante, le matériel de départ peut soit être un échantillon d’ADN issu d’un individu dont on souhaite connaître l’espèce, soit un échantillon d’ADN environnemental (eau, sol, fèces…) qu’on cherche à caractériser. Un échantillon d’ADN environnemental contient généralement des séquences issues de très nombreuses espèces, mais une seule est représentée ici, pour simplifier. Dans les deux cas de figure, l’ADN isolé est ensuite amplifié par PCR pour être présent en suffisamment grande quantité pour pouvoir être séquencé. L’amplification par PCR est réalisée grâce à des amorces spécifiques du gène utilisé comme code-barre. Le séquençage peut être réalisé avec la méthode de Sanger ou avec des méthodes de nouvelle génération (NGS) voire de troisième génération (TGS).
3. Une fois la séquence d’intérêt connue, il suffit de la comparer aux séquences de la base de données constituée à l’étape 1 pour identifier la séquence la plus ressemblante dans la base de données. Si celle-ci est suffisamment ressemblante (en général un seuil de similarité de 98 ou 99 % est retenu), cela permet d’identifier l’espèce à laquelle appartient l’ADN isolé à l’étape 2.
Source des images : Silhouette de chauve-souris :Ryan Kissinger pour NIAID NIH BioArt, domaine public ; Séquenceur de nouvelle génération : Ryan Kissinger pour NIAID NIH BioArt, domaine public ; Thermocycleur pour PCR : DBCLS, CC BY, Bioicons ; Base de données : gbrown, OpenClipart, CC0.
Alors qu’il avait été envisagé initialement d’utiliser le code-barre COI pour tous les Eucaryotes, il s’est avéré que s’il permettait effectivement de discriminer efficacement les espèces animales, même proches, son niveau de variabilité n’était pas adapté pour d’autres taxons. D’autres marqueurs sont alors utilisés : gène de la grande sous-unité de la Rubisco (rbcL) et gène de la maturase K (matK) chez les plantes, espaceurs internes transcrits (ITS) séparant les gènes des ARNr chez les champignons… 67 La base de données BOLD s’est donc adaptée, et inclut maintenant des collections de séquences de références basées sur différents fragments d’ADN, selon le taxon en question. Des bases de données existent également pour les bactéries et les archées comme SILVA. Dans ce cas, les codes-barres utilisés sont les gènes codant les ARN ribosomiques.
L’approche classique de DNA barcoding part du principe que l’échantillon prélevé sur l’organisme étudié ne contient que son propre ADN et, qu’après séquençage, une seule séquence code-barres sera obtenue. Il est cependant possible d’extraire un mélange de molécules d’ADN issues de différentes espèces et contenues dans un seul échantillon, comme des fèces, un contenu stomacal, un échantillon d’eau ou de sol, etc., par une approche de metabarcoding. Dans ce cas-là, après séquençage, plusieurs séquences codes-barres seront obtenues, et chacune pourra éventuellement être reliée à un nom d’espèce, permettant ainsi de connaître la composition en espèces de l’échantillon.
3. Identifier des individus appartenant à des espèces déjà décrites
Sur le papier, l’approche de DNA barcoding est donc très prometteuse. Rapide (les séquences d’ADN s’obtiennent maintenant en quelques heures), relativement peu coûteuse, elle ne nécessite pas d’être spécialiste du taxon pour identifier un spécimen. Par ailleurs, elle permet d’identifier un spécimen inconnu quel que soit son sexe, ce qui est particulièrement utile pour les espèces présentant un dimorphisme sexuel, ou son stade de développement, ce qui s’avère précieux pour les espèces à métamorphose. Les développements récents permettent également d’obtenir de l’ADN à partir de tissu ancien, mal conservé (ADN de Neandertal par exemple), et ouvrent ainsi un champ d’applications très vaste : biologie de la conservation, espèces invasives, ravageurs de cultures, santé publique (pathogènes, vecteurs…), espèces indicatrices de la qualité de l’environnement, agronomie, produits transformés pour la consommation, etc. Un exemple classique et représentatif se trouve dans une étude réalisée par Jacob Lowenstein et ses collaborateurs, dans laquelle les chercheurs ont échantillonné 68 sushis de thon dans des restaurants de Denver et de New York 8. Après séquençage, il s’est avéré que certains sushis ne correspondaient pas à l’espèce de thon annoncée, mais à une espèce protégée, alors que d’autres sushis n’étaient même pas au thon, mais à l’escolier noir (Lepidocybium flavorunneum), un poisson banni de la vente au Japon et en Italie en raison des violentes réactions gastro-intestinales qu’il peut provoquer…
Cependant, plusieurs limites existent à l’identification de spécimens par DNA barcoding. Tout d’abord, les bases de données sont loin de contenir des séquences pour toutes les espèces actuellement décrites. Par ailleurs, de nombreuses espèces vivant sur Terre sont pour le moment inconnues. Ensuite, l’utilisation d’un seul marqueur ADN constitue une source de biais qui peut induire en erreur le taxonomiste :
-
hétéroplasmie (présence d’un génome mitochondrial transmis par le père, en plus de celui transmis par la mère, chez certains bivalves par exemple) ;
-
pseudogène, c’est-à-dire une copie nucléaire non fonctionnelle du gène COI. Retrouvés chez les insectes par exemple, un tel pseudogène présente une séquence proche du gène COI mitochondrial et peut donc être amplifié par les mêmes amorces, puis séquencé. Heureusement, il est souvent possible de distinguer le pseudogène du gène COI mitochondrial, car le premier, qui n’est plus soumis à une pression de sélection, accumule souvent des codons stop.
-
transferts horizontaux de gènes entre espèces proches ;
-
tri incomplet des lignées, qui fait que la séquence d’un spécimen d’une espèce peut être plus proche des séquences de spécimens d’une autre espèce, que des séquences des spécimens de la même espèce (autrement dit, non-monophylie des espèces).
Une approche classique consiste alors à séquencer un autre gène, ou à prendre en compte d’autres caractères, mais cela devient alors plus coûteux, à la fois en argent et en temps, et les avantages de l’approche de DNA barcoding, simple, rapide et peu coûteuse, sont un peu perdus.
4. Découvrir et délimiter de nouvelles espèces
Rapidement après sa mise en œuvre, les taxonomistes utilisant les codes-barres ADN ont proposé que cette méthode n’était pas seulement intéressante pour identifier des spécimens inconnus, mais également pour mettre en évidence de nouvelles espèces, les délimiter et les décrire (voir par exemple 9). En effet, il n’est pas rare de mettre en évidence de nouvelles espèces potentielles lors de la création de la base de référence. En séquençant des spécimens issus d’expéditions de terrain ou de collections de muséums, il est très courant d’obtenir des séquences différant significativement des séquences d’espèces connues. Cela a conduit certains taxonomistes à décrire de nouvelles espèces sur la seule base d’un code-barres ADN.
Or la délimitation d’une espèce ne peut reposer sur un seul critère. Au contraire, pour délimiter une espèce, plusieurs propriétés doivent être testées. Il peut ainsi s’agir de propriétés biologiques (critère d’interfécondité), écologiques (partage d’une même niche écologique), morpho-anatomique (ressemblance physique), génétique (similarité du génome ou, plus spécifiquement, d’un code-barres ADN)… Cependant, ces propriétés seront vérifiées ou non selon le temps écoulé depuis la spéciation. Par conséquent, l’analyse de ces différentes propriétés, via les caractères génotypiques et phénotypiques, peut aboutir à des conclusions différentes. Il convient donc de confronter les hypothèses issues de l’analyse des différentes propriétés, de les interpréter pour comprendre quels sont les processus qui sont à l’origine de ces contradictions, pour finalement les intégrer, et aboutir à des hypothèses d’espèces plus robustes qu’en se fondant sur un seul critère. Cette démarche est celle de la taxonomie intégrative.
Dans cette optique, les codes-barres ADN constituent seulement un caractère parmi tant d’autres, que le taxonomiste va pouvoir utiliser pour proposer des hypothèses d’espèces. Comme ils sont faciles à obtenir, ils sont désormais souvent employés pour proposer des hypothèses primaires d’espèces, notamment dans les groupes mal connus, très diversifiés, et pour lesquels les caractères morphologiques sont difficiles à interpréter. Ces hypothèses primaires doivent ensuite être confrontées à d’autres caractères pour délimiter une espèce, dans une approche de taxonomie intégrative.
Au final, il est donc impératif de bien distinguer les utilisations qui peuvent être faites de la technologie de DNA barcoding. Collins et Cruickshank ont clarifié le débat, en distinguant deux tâches du taxonomiste : l’identification de spécimens, pour laquelle le DNA barcoding est un outil efficace et puissant, et la délimitation d’espèces, pour laquelle le DNA barcoding peut être utilisé pour proposer des hypothèses primaires 10. Ils ont ainsi banni l’utilisation du terme « identification d’espèces », un non-sens source de confusion, et pourtant encore largement utilisé dans la littérature en 2025.
Qu'est-ce qu'une espèce ?
Le débat entre partisans de plusieurs dizaines de concepts proposés dans la littérature semblait sans fin, jusqu’à ce que plusieurs auteurs, de Queiroz en tête, le clarifie 11121314. Ces concepts d’espèces (phylogénétique, écologique, biologique, etc.) ne sont en fait pas des concepts, dans le sens où ils ne définissent pas de façon universelle ce qu’est une espèce, mais plutôt des propriétés, qui sont ou ne sont pas vérifiées, selon l’avancement dans le processus de spéciation. Si ce processus de spéciation est terminé depuis longtemps, par exemple pour deux espèces ayant divergé depuis plusieurs dizaines de millions d’années, tous les « concepts », c’est-à-dire toutes les propriétés utilisées, aboutiront à la même conclusion : il s’agit bien de deux espèces distinctes. Par contre, deux populations qui viennent de se séparer se situent dans une « zone grise » : il ne s’agit plus vraiment d’une seule espèce, mais il ne s’agit pas non plus encore de deux espèces. Certaines propriétés seront vérifiées, d’autres non. Par exemple, morphologiquement, l’ours blanc est bien une espèce séparée du grizzly nord-américain. Biologiquement et génétiquement, ces deux formes appartiennent à la même espèce…
5. Conclusion
En parallèle de la clarification des concepts et des méthodes de la taxonomie, le DNA barcoding constitue un formidable outil d’analyse de la biodiversité. Cependant, le chemin vers un inventaire complet de la biodiversité est encore long, sans compter que dans le contexte actuel de la sixième crise d’extinction du vivant, un grand nombre d’espèces aura déjà disparu avant même qu’elles ne puissent être inventoriées. Malgré tout, le nombre d’espèces décrites chaque année augmente, mais faiblement (passant de 15 à 18 000 espèces décrites chaque année il y a 10-20 ans, à un peu plus de 20 000 de nos jours), tandis que la part des espèces décrites grâce, au moins en partie, aux codes-barres ADN, plafonne à 10 %.



